Fluorescence-lifetime imaging microscopy
Fluorescence-lifetime imaging microscopy or FLIM is an imaging technique for producing an image based on the differences in the exponential decay rate of the fluorescence from a fluorescent sample. It can be used as an imaging technique in confocal microscopy, two-photon excitation microscopy, and multiphoton tomography.
The lifetime of the fluorophore signal, rather than its intensity, is used to create the image in FLIM. This has the advantage of minimizing the effect of photon scattering in thick layers of sample.
Contents |
Fluorescence lifetimes
A fluorophore which is excited by a photon will drop to the ground state with a certain probability based on the decay rates through a number of different (radiative and/or nonradiative) decay pathways. To observe fluorescence, one of these pathways must be by spontaneous emission of a photon. In the ensemble description, the fluorescence emitted will decay with time according to
where
 .
.
In the above,  is time,
is time,  is the fluorescence lifetime,
is the fluorescence lifetime, 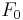 is the initial fluorescence at
is the initial fluorescence at 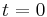 , and
, and 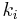 are the rates for each decay pathway, at least one of which must be the fluorescence decay rate
are the rates for each decay pathway, at least one of which must be the fluorescence decay rate 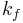 . More importantly, the lifetime, , is independent of the initial intensity of the emitted light. This can be utilized for making non-intensity based measurements in chemical sensing.
. More importantly, the lifetime, , is independent of the initial intensity of the emitted light. This can be utilized for making non-intensity based measurements in chemical sensing.
Measurement and processing
Fluorescence-lifetime imaging yields images with the intensity of each pixel determined by , which allows one to view contrast between materials with different fluorescence decay rates (even if those materials fluoresce at exactly the same wavelength), and also produces images which show changes in other decay pathways, such as in FRET imaging.
Pulsed illumination
Fluorescence lifetimes can be determined in the time domain by using a pulsed source. When a population of fluorophores is excited by an ultrashort or delta pulse of light, the time-resolved fluorescence will decay exponentially as described above. However, if the excitation pulse or detection response is wide, the measured fluorescence, M(t), will not be purely exponential. The instrumental response function, IRF(t) will be convolved or blended with the decay function, F(t).
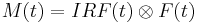
The decay function (and corresponding lifetimes) cannot be recovered by direct deconvolution using Fourier transforms because division by zero will produce errors and noise will be amplified. However, the instrumental response of the source, detector, and electronics can be measured, usually from scattered excitation light. The IRF can then be convolved with a trial decay function to produce a calculated fluorescence, which can be compared to the measured fluorescence. The parameters for the trial decay function can be varied until the calculated and measured fluorescence curves fit well. This process is known as reconvolution or reiterative convolution, and can be performed quickly by several software packages.
TCSPC
Time-correlated single-photon counting (TCSPC) is usually employed because variations in source intensity and photoelectron amplitudes are ignored, the time resolution can be upwards of 4 ps, and the data obeys Poisson statistics (useful in determining goodness of fit during reconvolution). More specifically, TCSPC records times at which individual photons are detected by something like a photo-multiplier tube (PMT) or an avalanche photo diode (APD) after a single pulse. The recordings are repeated for additional pulses and after enough recorded events, one is able to build a histogram of the number of events across all of these recorded time points. This histogram can then be fit to an exponential function that contains the exponential lifetime decay function of interest, and the lifetime parameter can accordingly be extracted.
Gating method
Pulse excitation is still used in this method. Before the pulse reaches the sample though, some of the light is reflected by a dichroic mirror and gets detected by a photodiode that activates a delay generator controlling a gated optical intensifier (GOI) that sits in front of your CCD detector. The GOI only allows for detection for the fraction of time when it is open after the delay. Thus, with an adjustable delay generator, one is able to collect fluorescence emission after multiple delay times encompassing the time range of the fluorescence decay of the sample.[1]
Phase modulation
Alternatively, fluorescence lifetimes can be determined in the frequency domain by a phase-modulated method. The intensity of a continuous wave source is modulated at high frequency, by an acousto-optic modulator for example, which will modulate the fluorescence. Since the excited state has a lifetime, the fluorescence will be delayed with respect to the excitation signal, and the lifetime can be determined from the phase shift. Also, y-components to the excitation and fluorescence sine waves will be modulated, and lifetime can be determined from the modulation ratio of these y-components. Hence, 2 values for the lifetime can be determined from the phase-modulation method. Consequently, if the lifetimes that are extracted from the y-component and the phase do not match, it means that you have more than one lifetime species in your sample.
Applications
FLIM has primarily been used in biology as a method to detect photosensitizers in cells and tumors as well as FRET in instances where ratiometric imaging is difficult. The technique was developed in the late 1980s and early 1990s (Bugiel et al. 1989. König 1989,[2] before being more widely applied in the late 1990s. In cell culture, it has been used to study EGF receptor signaling[3] and ErbB1 receptor trafficking.[4] FLIM imaging is particularly useful in neurons, where light scattering by brain tissue is problematic for ratiometric imaging.[5] In neurons, FLIM imaging using pulsed illumination has been used to study Ras,[6] CaMKII, Rac, and Ran[7] family proteins. FLIM has been used in clinical multiphoton tomography to detect intradermal cancer cells as well as pharmaceutical and cosmetical compounds.
FRET imaging
Since the fluorescence lifetime of a fluorophore depends on both radiative (i.e. fluorescence) and non-radiative (i.e. quenching, FRET) processes, energy transfer from the donor molecule to the acceptor molecule will decrease the lifetime of the donor. Thus, FRET measurements using FLIM can provide a method to discriminate between the states/environments of the fluorophore.[8] In contrast to intensity-based FRET measurements, the FLIM-based FRET measurements are also insensitive to the concentration of fluorophores and can thus filter out artifacts introduced by variations in the concentration and emission intensity across the sample.
References
- ^ Chang CW, Sud D, Mycek MA. Fluorescence lifetime imaging microscopy. Methods Cell Biol. 2007;81:495–524. pmid=17519182
- ^ Oida T, Sako Y, Kusumi A (March 1993). "Fluorescence lifetime imaging microscopy (flimscopy). Methodology development and application to studies of endosome fusion in single cells". Biophys. J. 64 (3): 676–85. Bibcode 1993BpJ....64..676O. doi:10.1016/S0006-3495(93)81427-9. PMC 1262380. PMID 8471720. http://www.biophysj.org/cgi/pmidlookup?view=long&pmid=8471720.
- ^ Wouters FS, Bastiaens PI (October 1999). "Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells". Curr. Biol. 9 (19): 1127–30. doi:10.1016/S0960-9822(99)80484-9. PMID 10531012. http://linkinghub.elsevier.com/retrieve/pii/S0960-9822(99)80484-9.
- ^ Verveer PJ, Wouters FS, Reynolds AR, Bastiaens PI (November 2000). "Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane". Science 290 (5496): 1567–70. doi:10.1126/science.290.5496.1567. PMID 11090353. http://www.sciencemag.org/cgi/pmidlookup?view=long&pmid=11090353.
- ^ Yasuda R (October 2006). "Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy". Curr. Opin. Neurobiol. 16 (5): 551–61. doi:10.1016/j.conb.2006.08.012. PMID 16971112.
- ^ Harvey CD, Yasuda R, Zhong H, Svoboda K (July 2008). "The spread of ras activity triggered by activation of a single dendritic spine". Science 321 (5885): 136–40. doi:10.1126/science.1159675. PMID 18556515.
- ^ The design of Forester (fluorescence) resonance energy transfer (FRET)-based molecular sensors for Ran GTPase, in press P. Kalab, J. Soderholm, Methods (2010), doi:10.1016/j.ymeth.2010.01.022
- ^ Wolfgang Becker, Axel Bergmann. Lifetime Imaging Techniques for Optical Microscopy. http://www.becker-hickl.de/abstracts/Lifetime%20Imaging%20Techniques%20for%20Optical%20Microscopy.htm